At age 2-1/2, Katie Lambertson of California was diagnosed with T-cell acute lymphoblastic leukemia (T-ALL), an aggressive condition marked by cancerous T cells crowding out healthy white blood cells in the bone marrow and blood. Over the next several months, Katie failed all standard therapies for her cancer, including three rounds of chemotherapy and a bone-marrow transplant involving total body irradiation. Her parents were told she had at most a month to live. Then came two lucky breaks that changed everything.
Katie’s father learned of a clinical trial evaluating an experimental drug developed specifically to kill the rapidly dividing T cells that characterize T-cell leukemia and other T-cell cancers. The drug, forodesine hydrochloride (Mundesine), was designed by Vern Schramm, Ph.D., professor of biochemistry and the Ruth Merns Chair in Biochemistry at Einstein. By targeting a specific enzyme called PNP, Mundesine caused uncontrollably dividing cancerous T cells—and only those cells—to be fatally overwhelmed by molecules that accumulate to lethal levels.
 Katie Lambertson as a 2-year-old and as a University of Colorado Boulder graduate. (Photos courtesy of Jeff and Linda Lambertson)
Katie Lambertson as a 2-year-old and as a University of Colorado Boulder graduate. (Photos courtesy of Jeff and Linda Lambertson)
As Katie’s experience shows, Mundesine can be miraculous. But with the drug’s wider use came a sobering observation: Only about 10% of patients treated with Mundesine experienced complete remission. The reason for the low response rate remained a mystery until a discovery reported in 2020 by British scientists.
Essentially, nonresponders have cancerous T cells containing high levels of a second enzyme, which digests the Mundesine-produced molecules, called dNTPs, that would otherwise kill them. The situation was different for the 10% of patients, such as Katie, for whom Mundesine was a lifesaver: their cancerous T cells had acquired a mutation that inactivated this second enzyme. With that enzyme absent, the dNTPs generated by Mundesine were able to wipe out cancerous T cells.
As a parent, I know it must be devastating for an incurable disease to strike your child. And as a basic scientist, nothing can compare to learning that a drug you’ve developed has saved a child’s life.
— Dr. Vern Schramm

Thanks to a five-year, $3 million National Cancer Institute grant awarded last year, Dr. Schramm is now designing inhibitors for SAMHD1. His Einstein collaborators on the grant are B. Hilda Ye, Ph.D., associate professor of cell biology, and Seiya Kitamura, Ph.D., assistant professor of biochemistry. Many researchers have tried—so far unsuccessfully—to inhibit SAMHD1. The desire to inactivate SAMHD1 is understandable, since doing so could yield several benefits beyond just treating T-cell cancers.
Helping against solid-tumor cancers: In several types of cancer (including pancreatic, liver, ovarian, and renal), high levels of SAMHD1 expression in tumor cells are associated with a poor prognosis. Some of those cancers could prove sensitive to a drug that reduces the SAMHD1 content of tumor cells.

Making cancer cells respond better to chemotherapy: As noted, most T-cell cancers don’t respond to Mundesine because SAMHD1 breaks down Mundesine’s killer dNTP molecules. SAMHD1, it turns out, also interferes with cancer chemotherapy. That’s because many chemotherapy drugs (e.g., cytarabine, clofarabine, fludarabine, and gemcitabine) work by forming anticancer agents very similar to the dNTP molecules produced by Mundesine. Just as Mundesine plus SAMHD1 inhibitors would boost dNTP to lethal levels in cancerous T cells, combining chemo with SAMHD1 inhibitors would make tumor cells more sensitive to chemo’s killing effects.

Enhancing longevity: At the ends of chromosomes are protective caps called telomeres, consisting of repetitive DNA sequences and proteins. Telomeres help keep the genome stable by preventing chromosomes from degrading or from fusing together. Every time a cell divides, its telomeres become slightly shorter. This telomere shortening contributes to aging, since it eventually renders cells incapable of dividing and causes cell death. In 2023, researchers reported that SAMHD1 activity restrains telomere length and, conversely, that depriving cells of SAMHD1 causes their telomeres to elongate, suggesting a possible role for SAMHD1 inhibitors in extending cell life. But caution would be needed, since excessive telomere elongation was found to make telomeres fragile.

SAMHD1, the enzyme now in Dr. Schramm’s crosshairs, is one of about 10,000 different enzymes contained in human cells. They act as catalysts, speeding the reactions needed for vital tasks such as converting food to energy and contracting muscles. But as exemplified by SAMHD1 and T-cell cancers, some enzymes also trigger health problems.
Countering them are enzyme inhibitors that constitute some 30% of all marketed drugs, including aspirin, ACE inhibitors for treating hypertension, and statins for controlling cholesterol.
Dr. Schramm and his colleagues have established an impressive track record for developing enzyme inhibitors—not only Mundesine but more than a dozen others as well. They have all resulted from Dr. Schramm’s great achievement: learning how to visualize, at the atomic level, the geometry of an enzyme’s transition state: a fleeting structure that lasts just a millionth of a billionth of a second and occurs during every enzyme reaction.
Once he has determined an enzyme’s transition state, Dr. Schramm works with partners to develop compounds that mimic it. These so-called transition-state analogs are designed to bind specifically to disease-causing enzyme molecules, permanently locking onto them so they can no longer cause problems. Such precise targeting also means that the chances of adverse effects—from attacking the wrong enzyme, for example—are practically nil.
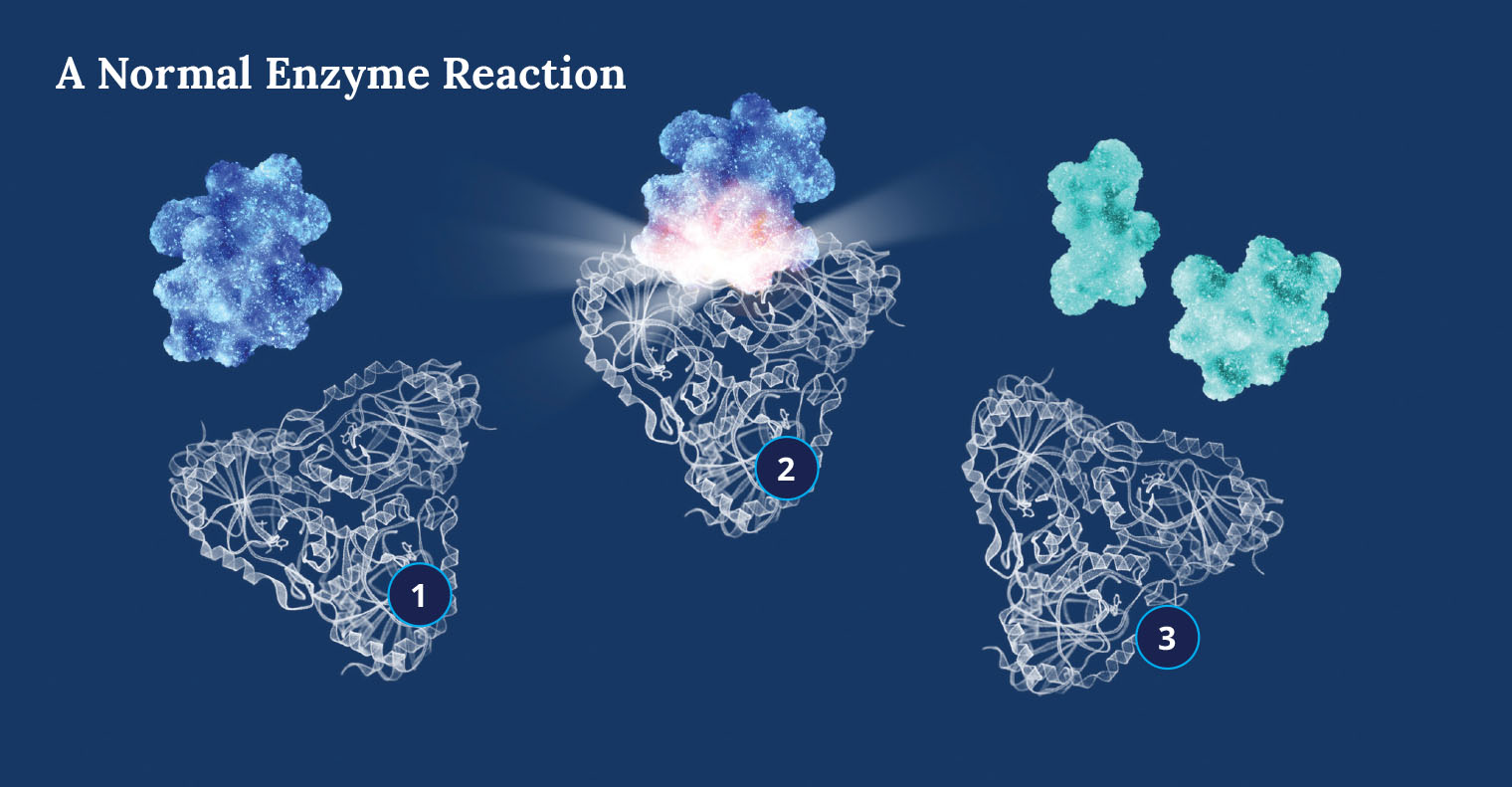 (1) An enzyme molecule’s active site is attracted to and binds with a substrate molecule (blue); a transition state then forms (2) to accelerate the substrate’s conversion into a product or products (3). (Illustration by Tatyana Starikova Harris)
(1) An enzyme molecule’s active site is attracted to and binds with a substrate molecule (blue); a transition state then forms (2) to accelerate the substrate’s conversion into a product or products (3). (Illustration by Tatyana Starikova Harris)
 Transition-state analogues inhibit the enzyme reactions that cancer cells, parasites and other disease-causing cells depend on. (1) The enzyme molecule is attracted to what appears to be a substrate molecule but is actually a transition-state analogue (blue-purple molecule); (2) the analogue binds the enzyme millions of times more tightly than a normal substrate molecule would, because the analogue’s bond-breaking energy is converted to binding energy; (3) with the enzyme’s active site permanently blocked by the analogue, it can no longer react with substrate molecules. (Illustration by Tatyana Starikova Harris)
Transition-state analogues inhibit the enzyme reactions that cancer cells, parasites and other disease-causing cells depend on. (1) The enzyme molecule is attracted to what appears to be a substrate molecule but is actually a transition-state analogue (blue-purple molecule); (2) the analogue binds the enzyme millions of times more tightly than a normal substrate molecule would, because the analogue’s bond-breaking energy is converted to binding energy; (3) with the enzyme’s active site permanently blocked by the analogue, it can no longer react with substrate molecules. (Illustration by Tatyana Starikova Harris)
While they haven’t yet developed SAMHD1 transition-state analogs, Dr. Schramm and his colleagues have achieved goal number one: figuring out the atomic structure of SAMHD1’s transition state. As with all the transition states that they’ve solved, doing so required computational quantum chemistry to search through thousands of SAMHD1’s theoretically possible transition states to find the models that most closely match experimental results obtained from kinetic isotope studies.
“With our blueprint of SAMHD1’s transition state in hand,” says Dr. Schramm, “we’re now designing stable molecular analogs that most closely mimic the transition-state structure and should—on paper at least—powerfully inhibit SAMHD1.” He will send the most-promising designs to colleagues in New Zealand, who will use them to synthesize actual transition-state analogs. Then will come lab testing to see which SAMHD1 analogs do best at inhibiting the enzyme in cancerous T cells. They’ll be tested against a novel Bronx-based, patient-derived cancer T-cell library.
“T-cell cancers disproportionately affect certain populations—not only the Japanese but also people from Caribbean countries such Haiti and Jamaica, who are highly represented in the Bronx,” says Dr. Schramm. “We’ve assembled a collection of cancerous T cells from patients in our borough and from Japan, and we’ll be testing our candidate SAMHD1 transition-state analogs against those cancer cells.”
The next step would be to test the SAMHD1 transition-state analogs in mice implanted with human T-cell cancers. If all goes well, the final step for obtaining U.S. Food and Drug Administration approval would be clinical trials to show that one or more of the SAMHD1 transition-state analogs are safe and effective in people with T-cell cancer.
“My involvement with T-cell cancers goes all the way back to the early 1990s, when I started working on the enzyme inhibitor that would become Mundesine, the drug that saved Katie’s life,” Dr. Schramm says. “If we can successfully develop an effective transition-state inhibitor for SAMHD1, I’m really optimistic that combining it with Mundesine will amount to a cure for most people who develop T-cell cancers. How rewarding that would be!”
For Katie Lambertson, the reward from Dr. Schramm’s lengthy quest has been the chance to live her life to the fullest. She says she has “pushed through” some health challenges following her treatment but has gone on to earn a bachelor of science degree in business and expects to receive a medical assistant diploma in June. Her next goal is to work at the same hospital where she was treated as a young child.
“It touches my heart to hear how my story has encouraged Dr. Schramm and other scientists at Einstein. I continue to be grateful for the vital work being done and hope my story can continue to inspire the persistence required to bring new life-saving therapies to people,” she says.
—With reporting by Sunita Reed
“Enzymologist Extraordinaire,” from the Summer/Fall 2018 issue of Einstein magazine, describes Dr. Vern Schramm’s journey to Einstein and his pioneering strategy for developing drugs.