
Error: No layouts found


In 2006, when Lindsy Osouna was on the cusp of her third trimester, prenatal testing showed that her daughter would be born with sickle-cell disease (SCD). “It was devastating,” she says, “but we decided that we have to face this.” After an intensive search, she and her husband, Kwasi, decided to entrust daughter Aniyah’s care to the SCD specialists at Children’s Hospital at Montefiore (CHAM), who collaborate with Einstein’s SCD researchers. These experts are focused on gaining insight into SCD and developing new therapies to transform the way the disease is treated.
Today, Aniyah is a charming and vivacious 11-year-old, healthy enough to attend school and even participate in gym class. But as with virtually all kids with SCD, her childhood has been far from easy, despite significant improvements in the care of children with the disease over the past few decades.
Kids with SCD now receive penicillin as standard treatment to prevent blood infections, a common and potentially fatal complication. Repeated blood transfusions can help keep anemia in check. And the drug hydroxyurea reduces the frequency of debilitating pain crises. As a result, the vast majority of children with SCD survive into their 40s—a significant improvement compared with the life spans of children born with SCD just a generation ago. But more advances are needed.
“For kids born with SCD, a minor cold or infection can turn into a major health crisis, and a case of the flu can be deadly,” says Henny Billett, M.D., professor of medicine and of pathology at Einstein and chief of the division of hematology at Einstein and Montefiore. “We’ve clearly got work to do to prevent these crises and to further extend the lives of SCD patients.”
Aniyah’s first crisis occurred when she was two and began gasping for breath. The Osounas rushed her from their home in Queens to CHAM, where they learned that her lungs were filling with fluid and verging on collapse due to acute chest syndrome, a potentially fatal pneumonia-like condition.
Her health stabilized after nine days, thanks to expert care from CHAM doctors and nurses—the type of care that should be available to all SCD patients but all too often is not. (See “Efforts to Increase Access,” below.)
Aniyah would experience the same emergency just a year later. Over the ensuing years came headaches, stomach cramps, gallstones, fevers, painfully swollen joints, an enlarged liver and early signs of retinal disease. In her short life, Aniyah has faced more health issues than many a frail octogenarian.
The sickle-cell center at CHAM is helping Aniyah and some 650 other children and adolescents with SCD overcome such challenges. It focuses on offering the best evidence-based treatments, recognizing preventable complications as early as possible, providing support for affected families and teaching its young patients how to manage their disease.

Deepa Manwani, M.B.B.S., with Lindsy Osouna and her children, Aniyah and Tristan.
Sickle-cell disease is a debilitating condition that affects approximately 100,000 Americans and occurs in about one in 365 African American births. The disease affects millions of people throughout the world, particularly those with ancestors from sub-Saharan Africa and Spanish-speaking regions of the Western Hemisphere (South America, the Caribbean and Central America).
People with SCD typically have inherited two mutated copies of the gene that codes for the beta subunit of hemoglobin, the protein in red blood cells that acquires oxygen from the lungs and carries it to tissues throughout the body. As a result, their hemoglobin differs by a single amino acid from “normal” hemoglobin. Individuals with a single errant copy of the gene carry the sickle-cell trait but do not develop SCD.
Blood cells normally are disc shaped, like a doughnut without a hole, which provides flexibility to the cells as they glide through blood vessels large and small. But in SCD, abnormal hemoglobin molecules aggregate to form filaments that distort red cells into their characteristic sickle shape.

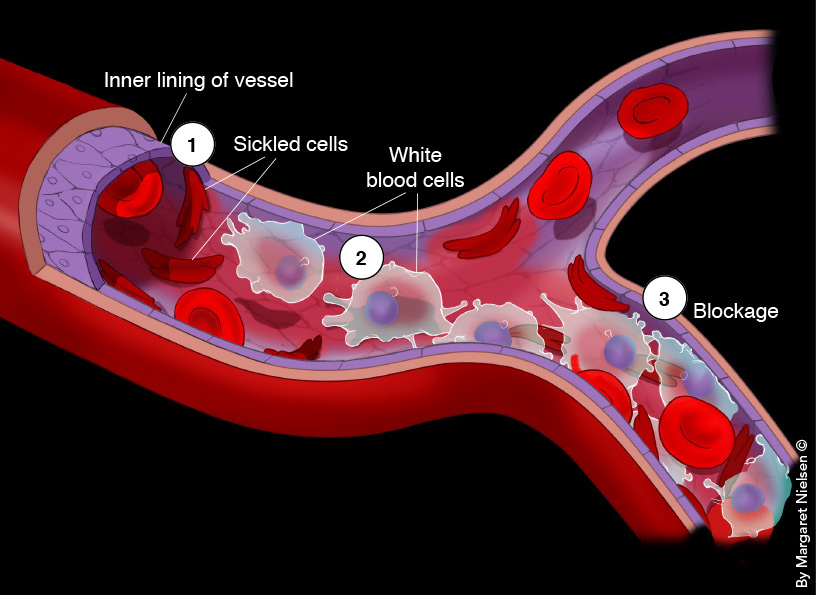
How vessels get clogged in SCD: Sickled cells irritate and inflame a blood vessel’s inner lining (1), which responds by producing cell-surface receptors called E-selectins that entrap white cells (2). Trapped white cells express activated Mac-1 adhesion molecules that bind sickled red cells, culminating in white cell/red cell clumps that hinder blood flow (3).
Sickled red cells are fragile and often burst, leading to anemia that reduces oxygen delivery to tissues. Even more important, their distorted shape makes the sickled cells sticky, causing vaso-occlusion—the clogging of small blood vessels that impairs oxygen delivery. Indeed, SCD is primarily a disease of oxygen starvation.
A chronic lack of oxygen causes organ damage that contributes to shorter life spans. Starting in early childhood, for example, SCD patients face a high risk of stroke due to faulty blood flow in the brain. (See “Silence of the Strokes,” below.) But long before such problems occur, oxygen deficits caused by SCD can cause excruciating pain.
“I get a lot of headaches and stomach pain,” says Aniyah matter-of-factly. “Sometimes my left arm hurts a little bit when I bend it.” If she is like most other SCD patients, the pain from too little oxygen nourishing her tissues is much worse than Aniyah lets on.
The scientific term for a painful red-cell clogging episode is a “vaso-occlusive crisis,” more commonly known as a sickle-cell crisis. It’s characterized by pain in the limbs, chest, stomach or lower back that can last from hours to weeks. Sickle-cell crises are the main reason that SCD patients require hospitalization. And over time, the crises contribute to the tissue and organ damage that impair quality of life and increase the risk of death for patients.
Patients experiencing sickle-cell crises must make do with supportive measures such as over-the-counter pain medications, heat compresses or massage, with a significant number of patients turning to potent painkillers such as opioids, putting them at risk for addiction. Pain that becomes chronic, as it often does in adulthood, is even harder to treat.
“Outside New York City and other major metropolitan areas, experts in sickle cell are hard to find,” says Suzette Oyeku, M.D., M.P.H., associate professor of pediatrics at Einstein and chief of the division of academic general pediatrics at CHAM. “Many primary care providers have never even seen a child with SCD. This is a travesty. Access to proper care shouldn’t be limited by your zip code.” Dr. Oyeku recently participated in the Hemoglobinopathy Learning Collaborative, a group of 15 institutions around the country that was formed to reduce disparities in SCD care. The collaborative is part of the Sickle Cell Disease Treatment Demonstration Program, funded by the federal Health Resources and Services Administration. “Clinical trials tell us what therapies are effective,” she says. “Our collaborative shows healthcare practitioners how they can implement these therapies in real-world settings.”
As an example, Dr. Oyeku cites the underused drug hydroxyurea, which, among other things, reduces the number of pain crises experienced by SCD patients. It works by reactivating the body’s production of fetal hemoglobin, which has an even higher affinity for oxygen than normal hemoglobin does. “But we’ve learned that many patients don’t take hydroxyurea because of misconceptions about the risks and benefits or because it wasn’t even offered to them,” she says. “In response, we’ve developed decision-making tools that encourage families and providers to have conversations about hydroxyurea. This approach, along with other strategies, has increased the proportion of patients who are taking the drug.” Other collaborative efforts include encouraging the use of telemedicine to bring hematology expertise to the hinterlands and developing expedited treatment protocols in emergency rooms to reduce the time that SCD patients must wait for pain relief.
“There’s a dire need for new therapies for sickle-cell crises,” says Deepa Manwani, M.B.B.S., who is Aniyah’s physician as well as a professor of pediatrics at Einstein and the director of pediatric hematology at CHAM. The program’s research arm is aimed at developing such therapies. One is a promising treatment for sickle-cell pain from the work of Einstein scientist Paul Frenette, M.D., professor of medicine and of cell biology and director of the Gottesman Institute for Stem Cell and Regenerative Medicine Research. Unlike opioid therapy used to dull pain, this approach instead blocks the underlying cause, which is vaso-occlusion.
In a 2002 PNAS paper, Dr. Frenette showed that sickled cells sticking to white cells and white cells sticking to the lining of blood vessels play a key role in vaso-occlusion. The process starts when sickled cells irritate and inflame the inner wall, or endothelial lining, of blood vessels, stimulating endothelial cells to produce cell-surface receptors called E-selectins. They entrap white cells, which in turn express adhesion molecules called activated Mac-1 that bind sickled red cells. The result: white cell/red cell clumps that cling to the lining of vessels and hinder blood flow (see illustration above).
To identify the specific adhesion molecules that cause cell clumping, Dr. Frenette added antibodies to blood to inactivate adhesion molecules one at a time and observe if vaso-occlusion occurred. Then he made an unexpected discovery: Unrelated antibodies he’d added for comparison purposes were preventing vaso-occlusion from occurring—suggesting that a widely available product rich in antibodies might help relieve sickle-cell crises.
The product, intravenous immunoglobulin (IVIG), is used to treat autoimmune diseases and has anti-inflammatory effects. Dr. Frenette’s research group gave IVIG to an SCD mouse model experiencing potentially fatal sickle-cell crises. A 2004 paper in Blood reported that IVIG dramatically inhibited interactions between red and white cells, increased microcirculatory blood flow and extended animals’ lives.
A phase 1 IVIG clinical trial directed by Dr. Manwani was completed in 2013. Children and adults admitted to CHAM and Mount Sinai for acute-pain crises were infused with varying doses of IVIG or a placebo solution. The trial yielded an IVIG dose that seemed well tolerated and effectively reduced activated Mac-1 adhesion molecules on white cells, making the cells less sticky.
In 2015, the National Institutes of Health (NIH) awarded Drs. Manwani, Frenette and colleagues a $1.6 million grant to conduct a phase 2 clinical trial assessing IVIG for treating sickle-cell patients eight to 21 years old admitted to CHAM with sickle-cell crises. The trial is assessing how well and how rapidly IVIG resolves pain crises and whether it helps reduce opioid use and the length of hospitalization.
Dr. Frenette’s cell-adhesion findings contributed to another promising therapy for SCD crises. In the 2002 PNAS study cited above, Dr. Frenette used a mouse model of SCD to show that P-selectin (a cell-adhesion molecule on endothelial cells) promotes vessel clogging by binding to white blood cells. Moreover, disrupting the expression of P-selectin on endothelial cells prevented binding from occurring and also prevented lethal vaso-occlusion.
Earlier this year, the New England Journal of Medicine published results of a multicenter, double-blind, placebo-controlled phase 2 trial in which 198 SCD patients received periodic infusions (14 per year) of a monoclonal antibody that neutralizes P-selectin. The treatment significantly reduced the number of pain crises compared with those reported by patients taking a placebo. Dr. Billett led the Montefiore portion of the study. A phase 3 clinical trial assessing anti-P-selectin antibodies (and again involving SCD patients cared for at Montefiore) is now under way.
In other sickle-cell adhesion work, Dr. Manwani is collaborating with Case Western Reserve University engineers to develop a way to measure the “stickiness” of red and white cells. “Our hypothesis is that acute vaso-occlusive pain is different from other types of pain commonly seen in SCD, such as neuropathy [nerve pain] and chronic pain,” explains Dr. Manwani. “If so, a marker for blood-cell adhesion would help us identify which patients would benefit from IVIG and other novel therapies, and perhaps allow us to predict and even prevent pain crises.” The researchers on this project are studying pediatric SCD patients being treated at Einstein-Montefiore.
Another Einstein-CHAM collaboration involves the surprising connection between the gut microbiome and the risk for a sickle-cell crisis.
Early in his career, Dr. Frenette discovered that vessel blockages in SCD occur when sickled red cells bind to white cells called neutrophils that have adhered to the vessel walls. He also noted that not all neutrophils were the same; some seemed inert, while others actively promoted inflammation—which is useful for attacking microbes but causes neutrophils to capture sickled red cells inside vessels.
In a 2015 paper in Nature, Dr. Frenette’s group reported that neutrophils become more pro-inflammatory as they age, in response to signals from the gut’s microbiome—the diverse populations of microorganisms in the intestines. Chemicals produced by the microbiome cross the intestinal barrier and enter the bloodstream, where they generate the old, overly active neutrophils that contribute to SCD crises.


Paul Frenette, M.D.
“Since the body’s microbiota seems to ‘educate’ neutrophils to age,” says Dr. Frenette, “we realized that purging those microbes with antibiotics might help against SCD.” As it turned out, administering antibiotics to SCD mice did appear to prevent sickle-cell crises; it markedly reduced interactions between neutrophils and red cells, resulting in increased blood flow and greatly improved survival of the mice.
“What was most exciting to us was the antibiotics’ beneficial effects on chronic tissue damage,” says Dr. Frenette. “Spleen enlargement in SCD mice was significantly reduced in the microbiota-depleted animals, and we also saw major reductions in liver damage. This is the first time that any drug therapy has been found to have an impact on the organ damage that can be so devastating in SCD.”
Were the findings in mice relevant to people? To find out, Drs. Frenette and Manwani obtained blood samples from SCD children being treated at CHAM and found that children taking penicillin had significantly fewer aged neutrophils than those not taking the antibiotic. “Daily penicillin for patients with SCD younger than five works really well in preventing infections,” says Dr. Frenette. “Our study suggests that antibiotics could potentially play an even broader role in benefiting older patients.”
Drs. Frenette and Manwani are planning a clinical trial to see if antibiotics can help SCD patients by preventing vaso-occlusive crises and the related long-term organ damage that shortens the lives of patients. Dr. Frenette is also working to identify which gut microbiome bacteria in particular are responsible for driving neutrophils to age.

When two SCD “carriers” like Lindsy and her husband Kwasi conceive, they have a three in four chance of having a baby free of the disease. The Osounas were counting on these odds when they decided to have a second child. “We also thought that if we had a healthy baby, we could bank the child’s cord blood and have a cure for Aniyah,” says Lindsy, referring to a stem-cell transplant.
Umbilical cord blood is rich in blood-forming, or hematopoietic, stem cells. Transplanting those stem cells into someone with SCD can provide a healthy blood supply, if the recipient’s marrow is first wiped out with chemotherapy. Such transplants are the only cure for SCD but are used sparingly because recipients and their donors—typically siblings—usually must have closely matching blood antigens, and risks for fatal complications are high.
Unfortunately, prenatal testing revealed that their new baby would also have SCD. “It was a big blow,” says Lindsy. “But again, we decided that we could deal with this.” The Osounas are not unusual in this regard. “Maybe 10 percent of parents will opt to terminate an SCD pregnancy based on genetic testing,” says Dr. Manwani. “It’s a very personal decision, guided by complex social and spiritual beliefs.”
Although Tristan, now six, has had fewer health problems than his sister, he struggles with severe asthma. “I’m sorry he’s not well,” says his mom, “but I’m happy he’s here.”
The Osounas take their children regularly to the CHAM sickle-cell center, despite the long drive from Queens. The family had considered moving to Nassau County, but didn’t want to change doctors. “I’m not sure I want to move farther away from CHAM,” says Lindsy. “It’s like a family there.”
Doctors are hoping that a stem-cell transplant may still be an option for both Aniyah and Tristan, even without a healthy sibling who can serve as a donor. CHAM’s bone marrow transplant program—one of the country’s largest—provides “haplo-identical” transplants that use partially matched family members (usually the mothers or fathers) as donors. Such transplants do involve serious risks, including infection and graft-versus-host disease. But they make cures at least theoretically possible for the majority of SCD patients.
Scientists recently developed a gene-editing technology called CRISPR that may offer a safer and more reliable SCD cure. CRISPR can cut out undesirable genes, or specific DNA sequences within genes, and splice in “normal” DNA—making it ideally suited for SCD, which results from a mutation involving a single nucleotide. Researchers have already used CRISPR to correct SCD mutations in animal models of the disease. A grant awarded to a Montefiore clinician and an Einstein basic scientist may bring CRISPR for SCD patients closer to reality.
Caterina Minniti, M.D., professor of medicine and of pediatrics at Einstein and director of the Sickle-Cell Center for Adults at Montefiore, and Eric Bouhassira, Ph.D., professor of cell biology and of medicine and the Ingeborg and Ira Leon Rennert Professor of Stem Cell Biology and Regenerative Medicine, received a $1 million grant from the Doris Duke Foundation’s Sickle Cell Disease/Advancing Cures program. Using blood and bone marrow samples from SCD patients, they’ll work to perfect CRISPR for editing the mutated hemoglobin gene in hematopoietic (blood-forming) stem cells. The ultimate goal: harvest an SCD patient’s hematopoietic stem cells, correct the defective hemoglobin gene and reinfuse the blood-producing cells into the patient. Depending on the percentage of normal blood cells produced, the procedure could reduce a patient’s symptoms or even provide a cure.

Ken Chen, Ph.D. (left), and Marc Vargas, B.S., use the CRISPR gene-editing technique in Einstein’s Transgenic Mouse Facility.
In caring for children who have SCD, the Osounas seek a healthy middle ground between helicopter and latchkey parenting. “We try to stress the importance of getting rest, staying hydrated, eating properly and taking their meds,” says Lindsy. As if on cue, Aniyah runs to the kitchen to retrieve her weekly pill organizer for an impromptu show-and-tell.
“We imagine there will be rough times ahead,” says their mom. “But we hope we’ve laid the foundation so that they will be able to take care of themselves later in life.”
When they turn 18, SCD patients “age out” of the nurturing world of pediatric care and must deal with the disease on their own. “For many young adults, it’s the first time they’ve had to make their own doctors’ appointments, and they have to get used to a whole new team of doctors and nurses,” says Dr. Minniti. “It’s a perilous time, when emergency room visits, hospital admissions and readmissions and even mortality rates increase greatly.”
Adult SCD patients may actually fare worse now than in the past. In a study published in 2013 in Public Health Reports, researchers used National Center for Health Statistics data to calculate SCD mortality rates (deaths per 100,000 African Americans) between 1979 and 2005. As expected, the mortality rate for children with SCD declined significantly over that time interval. But for adult SCD patients 19 and older, the mortality rate increased by one percent each year from 1979 to 2005. The authors, led by Einstein alum Sophie Lanzkron, M.D., ’91, M.H.S., attributed that rising mortality rate to “the lack of comprehensive care for adults with SCD.”
Caterina Minniti, M.D., with a patient, Joseph Peay.

At Montefiore, Dr. Minniti is working to eliminate the artificial transition from pediatric to adult care. “I’m a proponent of life-span care, with one group of physicians caring for patients throughout their lives,” she says. She worries that adult SCD patients in many parts of the country receive only episodic care, usually prompted by a sickle-cell crisis—a far cry from the care available to children.
“Visit pediatric clinics with their happy kids and then see how much worse the care is for adults with SCD,” says Dr. Minitti. “I encourage every pediatric hematologist to go to hospitals in their area to see what happens when their beautiful young patients become adults.”
When crises send SCD adult patients to the emergency room, “the focus is mainly on pain relief,” says Dr. Minniti. “But the pain, as bad as it may be, isn’t going to kill the patient. He is going to die because his kidneys are crashing or his heart is failing. Adult care must be multidisciplinary, because SCD causes complications that can damage all areas of the body, from the brain to the heart to the kidneys to the extremities.”
One of her own research interests is finding treatments for leg ulcers—painful and often disabling complications of SCD that affect 5 to 10 percent of all SCD patients and often take months or even years to heal. In July, the U.S. Food and Drug Administration awarded her a four-year, $1.9 million grant to carry out a phase 2 trial of a promising sodium-nitrite cream for treating leg ulcers in SCD patients.
In earlier work at the NIH, Dr. Minniti carried out a phase 1 trial of sodium-nitrite cream that found it was safe and well tolerated, and significantly reduced ulcer size and pain in the small number of patients treated. Applying the cream to the wound converts sodium nitrite into nitric oxide gas, which dilates vessels by relaxing their smooth muscle cells. This aids healing by increasing the amount of oxygenated blood that reaches the wound.

“We hope our phase 2 trial will confirm the promising results of our phase 1 study,” says Dr. Minniti. “No approved treatments for leg ulcers in SCD are available, and many patients with leg ulcers must now rely on chronic use of opioid drugs to relieve the excruciating pain that ulcers can cause.”
SCD-related problems tend to multiply and worsen with age. For example, years of working overtime to oxygenate the blood cause heart muscles to weaken and stiffen, setting the stage for heart failure and arrhythmias. Other chronic stresses compromise the joints, lungs, kidneys, gallbladder, spleen and brain.
“In treating SCD, we see all of the usual diseases of aging decades before they typically appear,” says Dr. Minniti. “Unfortunately, there’s only so much we can offer those patients. It’s hard to believe, more than a century after the disease was first characterized, that we have just two FDA-approved drugs. (Hydroxyurea and L-glutamine [approved in July 2017] both reduce the frequency of pain crises and the number of hospital visits needed to treat them.) In contrast, we have perhaps 25 drugs for HIV and 15 for cystic fibrosis.”
Nevertheless, Dr. Minniti is optimistic that things are heading in the right direction. “A number of drugs are now in clinical testing, including several at Einstein-Montefiore,” she says. “I think that in a few years we’ll have several therapeutic options. Then, perhaps, we’ll be able to move to the sort of combination therapy that transformed HIV from a deadly illness to a chronic one. If we can develop a cocktail of drugs that attack the different pathways leading to sickling, adhesion and vaso-occlusion—and if we can implement therapy early in life, before organ damage occurs—we will be able to extend and improve the lives of adults with sickle-cell disease.”
Tristan Osouna has high hopes that Dr. Minniti is correct. In a recent school assignment, he wrote: “The one thing in the world I would change is sickle-cell disease. I would change it by donating money to help hospitals get a cure. I have sickle-cell disease and I wish it would go away.”
Researchers at Montefiore and Einstein hope to make Tristan’s wish come true.
Strokes—both silent and overt—are extremely common among SCD patients, affecting between 25 and 40 percent of them. But why are SCD patients so susceptible to strokes?
Over the past four years Craig Branch, Ph.D., associate professor of radiology and of physiology & biophysics and director of the Gruss Magnetic Resonance Research Center at Einstein, has sought the answer through in-depth research on the biological mechanisms that underlie strokes in SCD. He is one of the only researchers to have used animal models of SCD to do so.
SCD-associated strokes are assumed to result from sickled cells clogging brain arteries and restricting blood flow, as occurs elsewhere in the body. But by studying mouse models of SCD, Dr. Branch found that brain blood flow associated with SCD strokes is actually unusually high—a finding recently confirmed in SCD patients studied in his laboratory and in others. Dr. Branch attributes this high blood-flow rate in the SCD brain to anemia and oxidative stress. But he has also observed a related alteration in brain metabolism never before seen, possibly in response to mild but chronic oxygen deficits in the brain.
“In SCD, the brain may be changing the way it manages its energy needs to reduce the likelihood of a major hypoxic event [a sudden loss of oxygen],” says Dr. Branch. “This compensation may unfortunately lead to increased blood flow in the brain, along with inflammation and a reduction in the brain’s ability to respond to conditions requiring increased oxygen—a combination that raises the risk of stroke.”
Today, physicians caring for SCD patients regularly assess their stroke risk by measuring blood-flow rate in the carotid artery; if the flow rate is abnormally high, they consider giving blood transfusions or treatment with hydroxyurea to increase the amount of oxygen reaching the brain. But confirmation of Dr. Branch’s studies could lead to other treatments—to reduce oxidative stress or improve oxygen delivery—that might ward off strokes among SCD patients.
Studies by Dr. Branch and Dr. Billett suggest that many SCD patients are experiencing silent, or subclinical, strokes that cause no obvious signs or symptoms. But silent does not mean harmless. “When we examine these SCD patients more closely,” he says, “we see clear evidence of cognitive deficits and brain pathology even though they’ve never been diagnosed as having stroke. Some of these findings may also be due to impaired metabolic brain processes.”
In a paper now under review for publication, Drs. Branch and Billett and a former Einstein medical student, Nicholas Farris, M.D., recruited adult sickle-cell patients who had no known history of stroke and an equivalent cohort of control individuals. All subjects underwent MRI brain scans as well as cognitive tests. The testing revealed that eight of 15 SCD patients had evidence of possible silent strokes and that all 15 SCD patients had significantly elevated brain blood flow and inflammation compared with the control subjects.




