
Error: No layouts found
More than one third of American adults—some 78 million people—are obese, and about 28 million have type 2 diabetes, which is usually associated with excess weight. Both problems persist despite numerous efforts aimed primarily at treating their “downstream” effects (e.g., liposuction to remove fat and type 2 diabetes drugs to alter insulin output by the pancreas).
Research by Einstein scientists suggests that treatment strategies should instead shift to the ultimate upstream target—the brain—since obesity and type 2 diabetes increasingly appear to be brain disorders. The Einstein research builds on decades of studies showing that the brain—the hypothalamus in particular—controls food intake and metabolism.
In 1940, Northwestern University Medical School’s A. W. Hetherington and S. W. Ranson published a classic paper in which they made lesions in the midregion of the hypothalamus of 22 rats. All but one of the rats increased their food intake and became obese. In a second landmark paper, in 1951, B. K. Anand and J. R. Brobeck of Yale University reported that lesions in a different part of the hypothalamus had the opposite effect: rats stopped eating and died of starvation.
These two studies, along with later research, have firmly established that the hypothalamus—an almond-sized portion of the brain—plays the central role in regulating both metabolism and weight.
But we now know that many more players get into the act as well.
When and how much we eat, how much we weigh and even whether we’ll develop type 2 diabetes depend on an extraordinarily complex web of interactions involving not only the hypothalamus but also the stomach, small intestine, pancreas, liver, blood, adipose (fat) tissue and even other parts of the brain, including the ventral tegmental area (part of the midbrain). To perform its finely calibrated job of controlling body weight and metabolism, the hypothalamus integrates messages from these other parts of the body and responds with its own directives (in the form of hormones, neurotransmitters and activated signaling pathways) that control how much we eat and how much energy we expend.
Molecule by molecule, Einstein scientists are trying to understand how the body controls its weight—and what goes wrong when this finely tuned regulatory mechanism breaks down. Their detective work has revealed several molecular glitches—in the hypothalamus and elsewhere—that lead to weight gain and elevated glucose levels that occur in type 2 diabetes. Repair of those glitches may lead to treatments for obesity and diabetes that target the root causes of these problems.

Streamson C. Chua, Jr., M.D., Ph.D.
Gastric bypass surgery has helped countless people shed excess pounds and control type 2 diabetes. But this operation—which shrinks the size of the stomach and reroutes nutrients to the lower intestine—is far from perfect. Many patients eventually regain much of their lost weight, and the surgery itself has a sobering mortality rate, with about one out of every 200 patients dying from it.
What if a pill could mimic the positive effects of gastric bypass surgery and avoid the negative ones? Sounds far-fetched, but research into this intriguing idea is well under way in the laboratory of Streamson C. Chua, Jr., M.D., Ph.D., a professor in the department of medicine (endocrinology) and in the Dominick P. Purpura Department of Neuroscience.
Throughout his career, Dr. Chua has concentrated largely on the genetics of obesity and diabetes and on leptin (the “satiety hormone”). Gastric bypass didn’t cross his mind until a colleague studying its physiological effects sought his help regarding a protein called fibroblast growth factor 19, or FGF19.
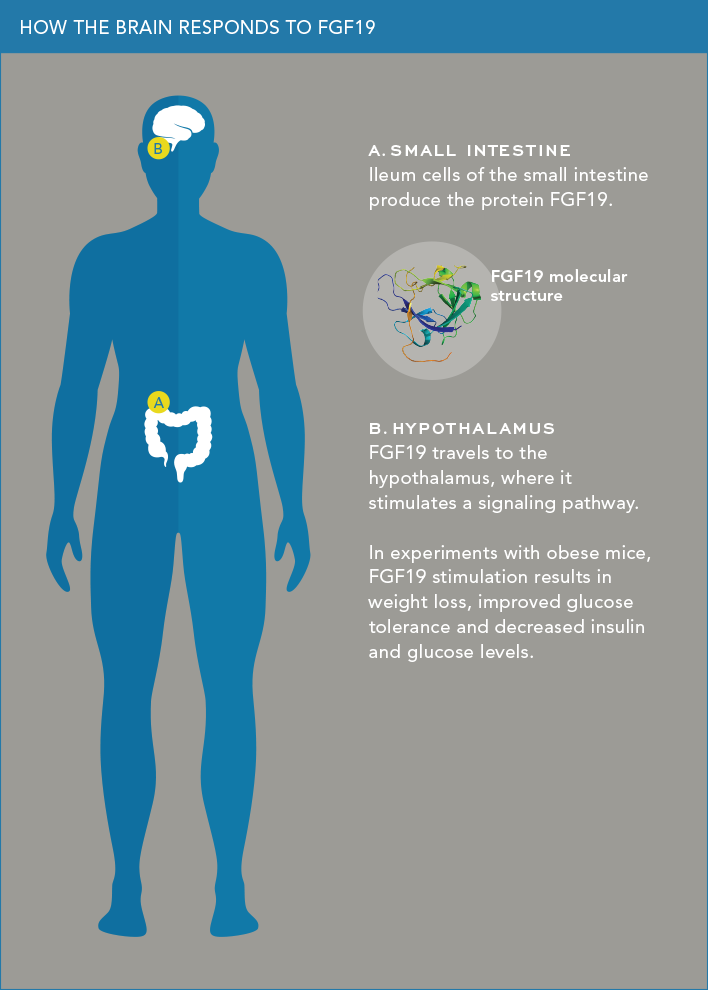
“It turns out that you can replicate the weight-loss effects of gastric bypass with a simpler and less-risky procedure called a biliary bypass,” says Dr. Chua. “In this procedure [typically used when a tumor has blocked the bile duct’s entry to the small intestine], the bile duct from the gall bladder is surgically relocated to a lower section of the small intestine called the ileum. This anatomical change stimulates bile acid receptors in the ileum, which in turn trigger the ileum’s absorptive cells, known as enterocytes, to produce FGF19.”
Dr. Chua’s colleague wanted to know what FGF19 is doing. “Everyone thought that FGF19 mainly works peripherally, in the liver and in brown fat,” says Dr. Chua. “But some evidence suggested it might also be affecting the hypothalamus,” the brain’s appetite-control center.
Dr. Chua found in mouse studies that the brain responds when FGF19 is administered in the peripheral circulation. He next injected FGF19 directly into the hypothalamus of genetically obese mice and of mice fed a high-fat diet. In both cases, the injected protein caused weight loss by selectively stimulating a signaling pathway called ERK1/2 and thereby silencing AGRP neurons (which promote feeding and insulin secretion). The FGF19-treated obese mice also exhibited several healthy signs—improvements in glucose tolerance and insulin sensitivity along with decreases in both insulin and glucose levels, as Dr. Chua reported in 2013 in Molecular Metabolism.
FGF19 would seem to be an ideal weight-loss drug. But unfortunately, it causes liver cells to proliferate. So over the last two years, Dr. Chua has been trying to identify peptides (small chains of amino acids) that duplicate FGF19’s actions.
Working in collaboration with the Danish pharmaceutical company Novo Nordisk, he has already identified several promising drugs that bind to FGF19-related receptors on AGRP neurons and silence their activity. Dr. Chua now plans to test these peptides in obese mice and glucose-intolerant mice to see if the peptides work as predicted in reducing blood-glucose and insulin levels.

Young-Hwan Jo, Ph.D.
Leptin is called the “satiety hormone” because it plays a key role in curbing appetite. Fat cells that have grown suitably plump synthesize leptin and release it into the bloodstream. The hormone travels to the brain’s hypothalamus—telling us, in effect, “You’ve had enough food, so step away from the table.”
But for reasons not fully understood, leptin’s communication with the brain starts breaking down when people gain excess weight. As the pounds accumulate, the brain becomes less sensitive to leptin’s “stop eating” message. So overweight people overeat, making a bad situation worse.
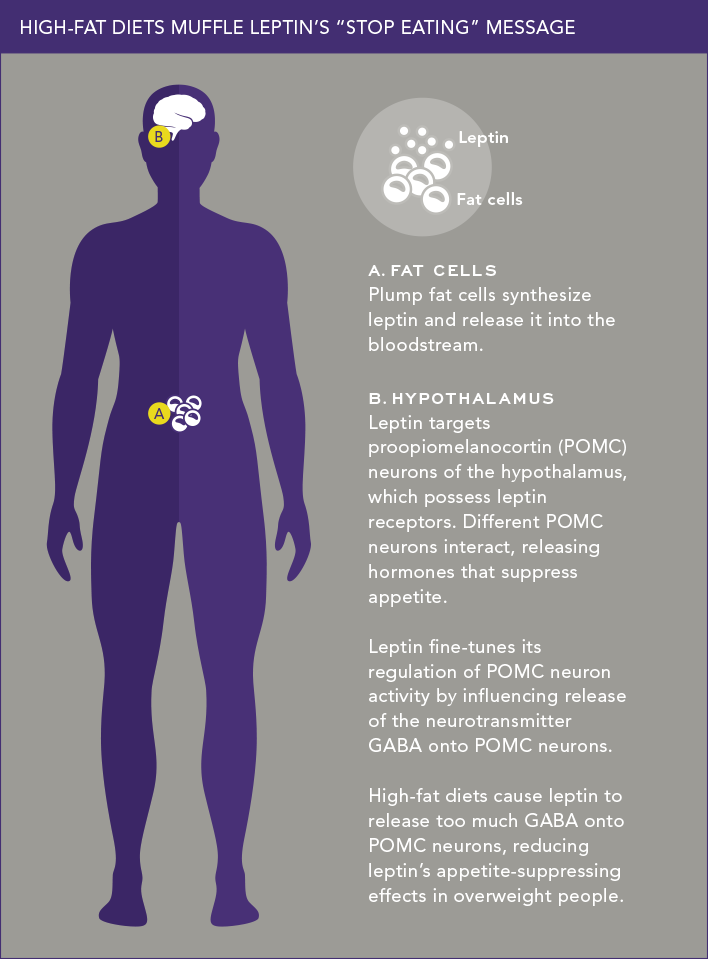
With support from a five-year, $1.82 million grant from the National Institute of Diabetes and Digestive and Kidney Diseases, Young-Hwan Jo, Ph.D., is trying to determine why for so many people leptin no longer works, the body’s energy balance breaks down and obesity results. Leptin has a special affinity for a bundle of hypothalamic neurons called the arcuate nucleus (ARC), which is crucially important for maintaining a neutral energy balance (in which energy intake equals energy expenditure). Within the ARC are thousands of proopiomelanocortin (POMC) neurons sprouting numerous leptin receptors.
These POMC neurons were long thought to be identical. But recent studies by Dr. Jo and others have found at least two distinct types. “We hypothesize that the different types of POMC neurons have different functions and that their interplay regulates energy balance,” says Dr. Jo, an assistant professor of medicine (endocrinology) and of molecular pharmacology. “In response to leptin and other signaling molecules, the different POMC neurons interact to release hormones needed to suppress appetite.”
Leptin molecules activate leptin receptors on POMC neurons, causing them to release melanocortin peptides as well as neurotransmitters that powerfully inhibit food intake. “The different types of POMC neurons seem to maintain energy balance by coordinating the release of these appetite-suppressing chemicals,” says Dr. Jo. “We propose that being overweight somehow disrupts this highly orchestrated interaction among different POMC neurons.”
Dr. Jo’s work has already revealed one possible glitch. Leptin, it turns out, not only turns on POMC neurons and their anti-appetite message; it also fine-tunes POMC activity by either (1) causing other neurons to release GABA (the central nervous system’s main inhibitory transmitter) onto POMC neurons or (2) preventing those neurons from releasing GABA. Whether leptin inhibits or activates GABA’s release onto POMC neurons depends, in turn, on signals from nutrients.
In a study in 2015 in Nature Communications, Dr. Jo and colleagues showed that glucose levels in the blood influence leptin’s action in releasing GABA. When glucose levels are high (as might occur following a meal), leptin inhibits GABA release from neurons (meaning that the POMC neurons’ “stop eating” message is unimpeded). Conversely, at low blood-glucose levels, leptin stimulates GABA release—in effect curbing POMC neuron activity and encouraging eating when blood sugar is low. But more interesting was what happened in mice that had been fed a high-fat diet for three weeks.
“We assumed that elevated body weight in these mice would make leptin inhibit GABA release onto POMC neurons, allowing their ‘stop eating’ message to be broadcast,” says Dr. Jo. “But we made the unexpected finding that the high-fat diet actually prompted leptin to enhance GABA release, which paradoxically may reduce leptin’s appetite-suppressing effects in obese animals. This finding may explain the energy-balance breakdown that occurs when people become overweight.”
Dr. Jo’s further research into POMC neurons may lead to strategies for helping their “stop eating” message prevail over signaling molecules trying to squelch it.

Meredith A. Hawkins, M.D.
Diabetes by definition involves abnormally high glucose levels in the blood. Those high levels eventually damage tissues, leading to the serious complications related to diabetes. While we tend to associate elevated blood glucose with too much dietary sugar and other carbohydrates, the main source of hyperglycemia in type 2 diabetes is actually excessive endogenous glucose production—chiefly by the liver, which produces, stores and secretes glucose into the bloodstream.
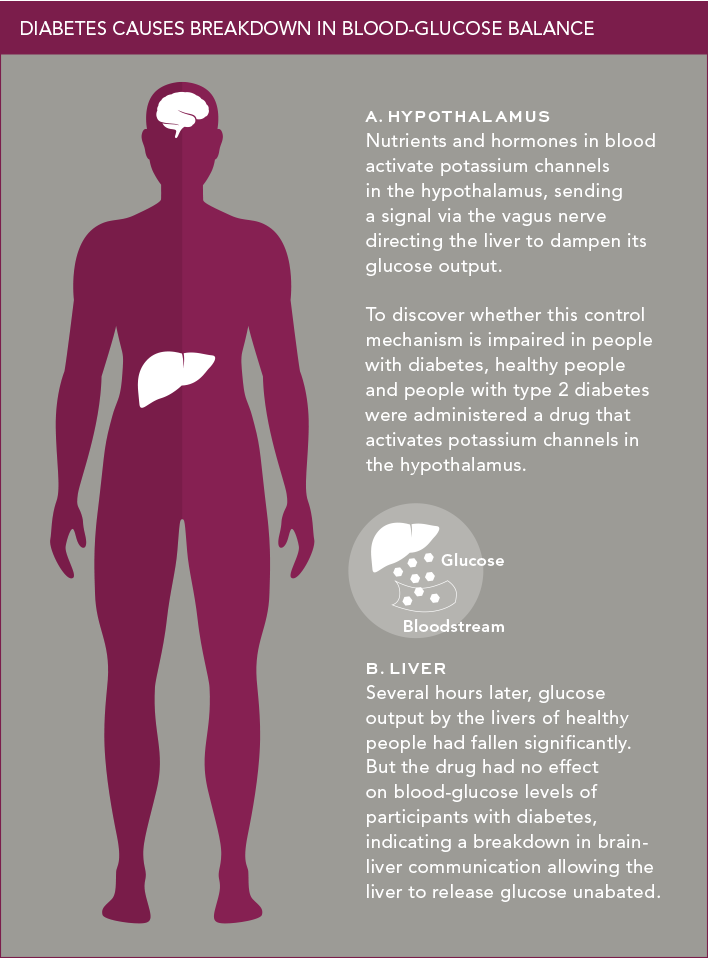
Over the past decade, professor of medicine Meredith A. Hawkins, M.D., and her Einstein colleagues have shown that the hypothalamus plays a key role in regulating blood-glucose balance in healthy people. The researchers found that when the hypothalamus detects certain nutrients and hormones in the blood, potassium channels in the hypothalamus become activated; these activated channels send a signal via the vagus nerve that tells the liver to dampen its output of glucose to the bloodstream.
Dr. Hawkins, who is associate director of the Einstein−Mount Sinai Diabetes Research Center (DRC) and director of Einstein’s Global Diabetes Institute, recently investigated whether this glucose-regulatory mechanism is defective in people with diabetes, which could help account for their abnormally high blood-glucose levels.
“Despite all the medications available for lowering blood-glucose levels, half of patients with type 2 diabetes are not able to achieve adequate blood-sugar control,” she notes. “Given the role of the hypothalamus in controlling blood glucose in healthy individuals, we looked at whether this control mechanism is impaired in people with diabetes. If so, then therapies could possibly be developed to restore the brain’s regulation of blood-glucose levels.”
To two groups of people—seven healthy individuals and eight with moderately to poorly controlled type 2 diabetes—Dr. Hawkins and her team administered oral diazoxide, a drug that activates potassium channels in the hypothalamus. (Diazoxide is used clinically to inhibit the pancreas from secreting excessive amounts of insulin; in this experiment, the researchers controlled insulin secretion by the pancreas to ensure that any change in blood-glucose levels would occur only through diazoxide’s effect on the brain.)
Several hours after diazoxide was given, blood tests showed that the drug had significantly lowered the glucose output of the healthy participants’ livers by an average of 27 percent—showing that the “activated potassium-channel pathway” of the hypothalamus operated normally to control blood-glucose levels. By contrast, diazoxide had no effect on the blood-glucose levels of participants with diabetes, indicating a breakdown in communications between the brain and liver that allowed the liver to release glucose unabated. The findings were published in May in Diabetes.
“My colleagues and I are now trying to understand the various components of this signaling system and how we might repair it when it’s not functioning normally,” says Dr. Hawkins, who also holds the Harold and Muriel Block Chair in Medicine and is an attending physician in the department of medicine’s endocrinology division at Montefiore.

Gary J. Schwartz, Ph.D.
Second-generation antipsychotic drugs—brands including Abilify, Risperdal, Seroquel and Zyprexa—debuted in the 1990s. They help millions of Americans cope with psychiatric disorders such as schizophrenia, depression and anxiety. But these medications (sometimes referred to as atypical antipsychotic drugs, or APDs) also cause adverse metabolic side effects, including weight gain and increased risk for type 2 diabetes.
Little is known about how APDs disrupt the body’s metabolism, and even less about how to prevent the problem. Gary J. Schwartz, Ph.D., a professor of medicine (endocrinology) and of neuroscience, is among a handful of researchers trying to find some answers, thanks to a three-year, $2.5 million grant from the Department of Defense.
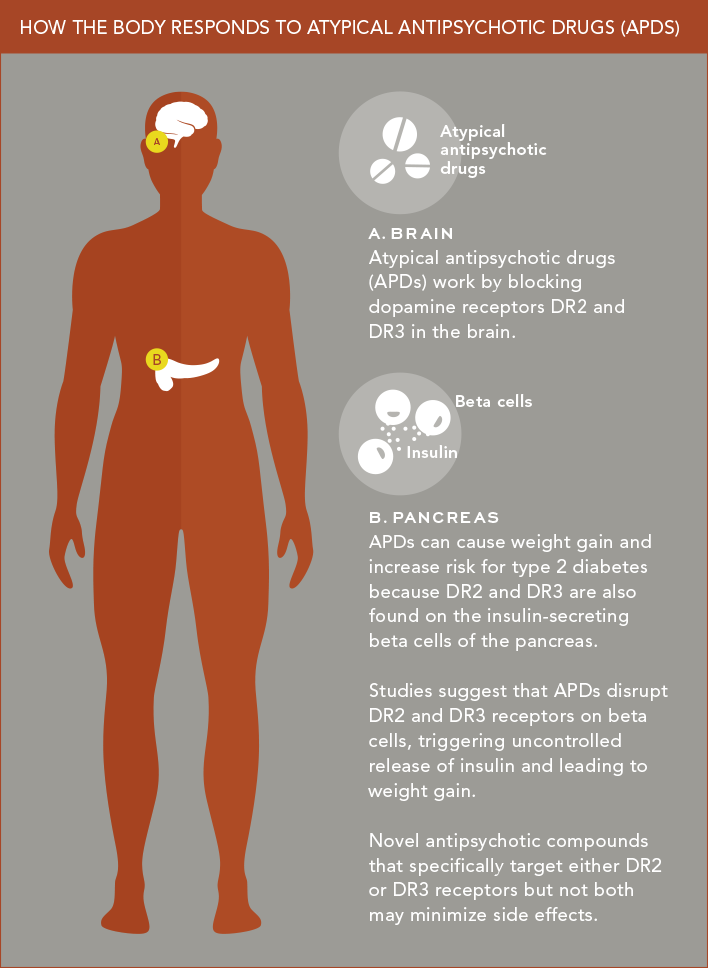
Schizophrenia and several other psychiatric illnesses are characterized by an overabundance of the neurotransmitter dopamine in the central nervous system. APDs work by countering this dopamine excess. They do so in part by blocking two types of dopamine receptors (called DR2 and DR3) that dopamine activates.
The problem, says Dr. Schwartz, is that DR2 and DR3 are also found outside the brain—most importantly, on insulin-secreting beta cells of the pancreas. Studies by Dr. Schwartz and colleagues at Columbia University Medical Center suggest that APDs disrupt these DR2 and DR3 receptors, triggering uncontrolled release of insulin from beta cells and leading to weight gain.
Dr. Schwartz is trying to disentangle the relative “contributions” that pancreatic beta cells’ DR2 and DR3 receptors make to APD-induced metabolic problems. He is also teasing out the relative importance of DR2 and DR3 receptors in causing these problems. To do so, he and his colleagues are developing “knockout” mice with pancreases lacking either DR2 or DR3 as well as novel antipsychotic compounds that specifically target either DR2 or DR3 receptors, but not both.
“We hope this work will lead to new antipsychotics with fewer metabolic side effects, which in turn would improve treatment compliance,” says Dr. Schwartz. The findings, he adds, may also reveal new strategies for treating overeating, weight gain and impaired glucose tolerance.
The Defense Department has a keen interest in the topic. Several hundred thousand veterans currently take APDs for post-traumatic stress disorder. Their prevalence of metabolic problems is more than twice that of the general population.

Dongsheng Cai, M.D., Ph.D.
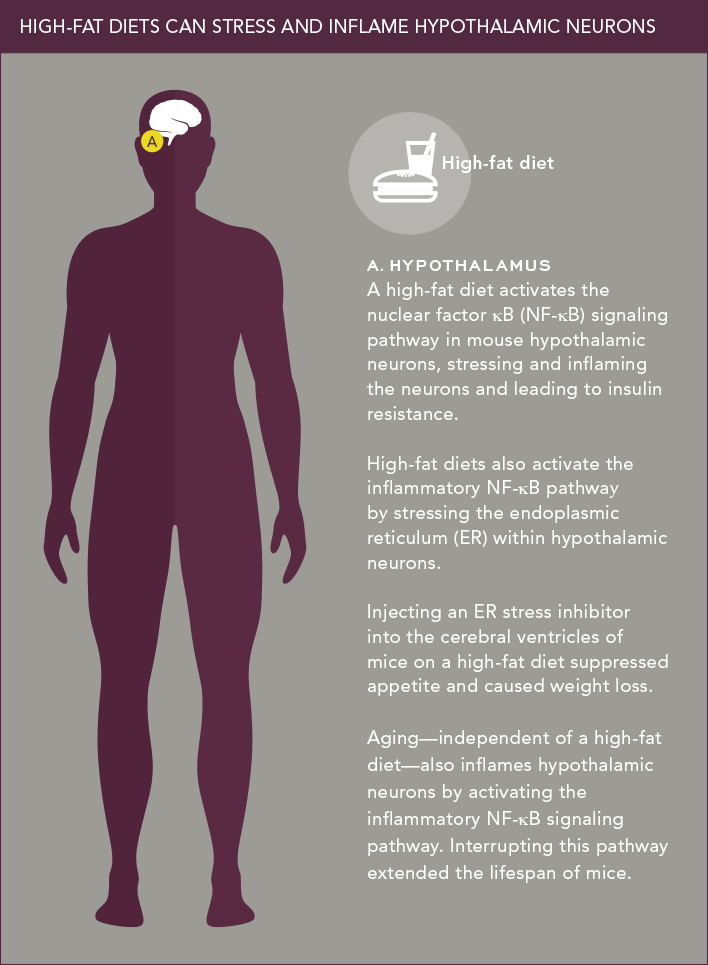
It’s well known that stress contributes to many health problems, from headaches to hypertension; it also appears to play a role in obesity and diabetes. It’s not the stress we associate with a tight deadline at work, but rather intracellular stress caused mainly by nutrient overload. Recent studies show that intracellular stress can trigger damaging inflammatory responses inside neurons of the hypothalamus.
Dongsheng Cai, M.D., Ph.D., a professor of molecular pharmacology, was one of the first researchers to connect hypothalamic inflammation to metabolic dysfunction. Studies had shown that overeating can chronically inflame certain peripheral tissues and lead to insulin resistance—an early step in type 2 diabetes. About a decade ago, Dr. Cai reasoned that something similar might be happening in the hypothalamus.
Hundreds of molecules play a role in inflammation, so deciding on possible culprits wasn’t easy. Dr. Cai hypothesized—correctly, it turns out—that a protein complex called nuclear factor κB (NF-κB) was a prime suspect. “Many molecules are involved in intracellular inflammation, but NF-κB sits at the center of this regulatory map,” he explains.
In studies funded by the National Institute of Diabetes and Digestive and Kidney Diseases, he found that the NF-κB inflammatory signaling pathway is present in the hypothalamic neurons of mice, and that a high-fat diet turns it on. He then showed that interrupting this pathway—by knocking out the gene for IKKβ, an enzyme that activates NF-κB—suppresses inflammation in hypothalamic neurons and also stops the animals from overeating and becoming obese or diabetic.
In other mouse experiments, Dr. Cai found yet another way that a high-fat diet triggers the inflammatory NF-κB signaling pathway: by stressing the endoplasmic reticulum (ER)—a membranous structure involved in protein and lipid transport—within hypothalamic neurons. Injecting the ER stress inhibitor tauroursodeoxycholic acid into the cerebral ventricles of mice on a high-fat diet suppressed their appetite and reduced their degree of obesity.
Today, Dr. Cai is exploring how to apply these findings clinically. “We have a number of ideas for selectively blocking this pathway in the hypothalamus using peptides,” he says.
In a fascinating related study involving mice, Dr. Cai recently investigated whether aging might cause weight gain and diabetes through its effects on the hypothalamus. He found that aging—independent of a high-fat diet—does indeed trigger inflammation within neurons of the hypothalamus by activating the NF-κB pathway. Dr. Cai was able to extend the life span of mice by interrupting this pathway. Together, these discoveries point to an entirely new approach for controlling metabolic disorders as well as certain aging-related health problems.
The efforts of these and other Einstein researchers have clearly shown that obesity and diabetes can be viewed as brain disorders. Progress in identifying the specific molecular defects and neuronal signaling pathways involved is already shedding light on potential new therapeutic strategies. Yet much remains to be learned about the complex metabolic integration that controls glucose metabolism and energy balance in the body.
“Studies focusing on the brain—and on discrete components of the hypothalamic signaling network in particular—may soon get a boost here, thanks to the strategic plan that Einstein announced in June,” says Jeffrey E. Pessin, Ph.D., DRC director and the Judy R. and Alfred A. Rosenberg Professorial Chair in Diabetes Research. The plan’s six priority research areas include the Brain Science Initiative as well as “obesity and metabolic disorders,” including diabetes.




